Nature Communication 发文 《
Extracting medicinal chemistry intuition via preference machine learning》,译文《偏好学习提取药物化学性质》。
本文主要由 Jingyi 翻译,原文参考《
Nature子刊:机器学习利用人类化学家直觉助力药物发现》,作者 生物世界。
01 文章摘要
发现新药工作中,“先导优化”是一项艰巨的工作。许多药物化学家所学的,其实是被权衡过的,为了达到一种所需的分子性质档案。建树起专业知识,以协同地成功驱动此类项目,是非常耗时的过程,通常会占据一个化学家职业生涯好几年的时间。这篇论文,我们打算使用 AI 来替代这一过程,即“学习即排序”技术(learning-to-rank)。“学术即排序”技术从以下历时几个月的过程获得反馈,这些工作由 35 位来自诺华的化学家完成。针对习得的、可触达的常任务的有规用性,我们会展示例子,如化合物优先顺序、基序合理化和有偏见的从头药物设计。提供了带注释的响应数据,并通过许可的开源许可证提供了开发的模型和代码。
an arduous endeavour 一项艰巨的工作
medicinal chemist 药物化学家
a desired molecular property profiles 一种所需的分子性质档案。这里指的是,人们所期待的分子性质所显示出期待中的样子。
successfully drive such projects collaboratively 一起成功驱动此类项目
typically spans many years within ... 通常占据…好几年的时间
over the course of several months 几个月的工作
routine tasks 常规任务
lead optimization 先导化合物,药物化学术语。
02 问题介绍
“药物发现”是复杂又多工序的过程,并且需要掌握化学、生物子学科交界面的技能。在好几道工序中,特别是先到优化步骤,药物化学家——湿实验室或计算——发挥着核心作用。由于药物化学家常规任务就是识别,哪些物质组成化合物,以及评估之后的轮数。为了达到这些,药物化学家经常需要重新审视数据,包括复合性质,例如活性、ADMET、目标结构信息,还有很多其他性质指标。因此,成功发新药过程,不仅仅基于高质量产出的实验数据,更是最终稳定而有效的决策,由药物化学家团队作出的决策。
在职业生涯镇南关,药物化学家建立其专业知识,使其能更有效够做出上述决策(例,复合优先级),这意味着他们必须对相关要素有知觉,即使得之后可成功迭代的复合物,这些迭代将逐渐形成早期药物发现过程。尽管以前曾试图用基于规则的方法将这些知识规范化(如,结构化警报),或简单的化学信息可用性评分(例,药物相似性),以捕捉到化学物质涉及排序能力的微妙性和复杂性,这些对于化学家而言,仍然都是基本挑战。考虑到这一冬季,这项工作中,我们研究了部分上述知识,是否可以提炼成机器学习模型。这类模型未来期望能够成为一种辅助手段,助力于先导优化或者其他药物发现决策过程,和其他在行业内以及发布的推荐系统一样。
鉴于目前药物化学家目前大多数都是人工工作,因此难以避免倾向于主观偏差。有好几篇文献都评估了,药物化学家倾向于同意自己的想法到什么程度,以及他们同事的结论。本篇论文大多数工作聚焦于,向化学家展示一系列复合物,这些复合物在之后几轮工序中会被逐一筛选。上述工作目的是,评估化学家选择的(复合物)是否会与其同事的选择重合,以及化学家们的优先选择是否具有一致性。本报告总的来说,并不关注化学家之间的共同结论,更多关注不同结论,这些结论通常和一些心理因素联系在一起,例如损失厌恶。另一篇更接近 Nature 的文献,也会在本文提及,评估一小部分化学家是否能依据一些性质来评估复合物,例如,Likert Scale(测量心理因素的工具)、或者药物相似性(drug-likeness),然后根据反馈训练机器学习模型。Merck 最近的研究也用了同意的衡量方法,对分子复杂性的内部众包代理进行建模。相比上述两篇文献的模型输出结果,发现相差不大,也未作过多调整,发表的论文已然倾向于锚定心理因素,该因素会使得决策(复合物)过程受到特定心理条件、特定场景条件影响。最近一项工作使用了类似实验方法,在本文中也提到,他们使用了多孔有机笼子(方法)。
本篇我们试图克服上述的限制,采取的方式也是非常有名的多玩家设定。我们设定目标就是排序分子(注,排序复合物,用于选择先到化合物),并将该任务抽象成偏好学习问题,展示个人喜好,通过配对来识别个人喜好,基于简单神经网络模型框架。基本机制,简单来说,基本思想如图1所示,证明概念的数据收集轮数,处理之后输出评估结果,即提出的研究及方法是否成功地克服了前文提到的认知偏差限制。35名诺华的化学家参与此次研究(用 wet-lab,或者计算,或者分析方法),收集了超过 5000 个注解,在前述好几道工序(或轮数)中,由主动学习方法主导这部分研究。我们展示了模型输出的非显性评分函数,捕捉到目前化学家用计算机度量化学信息所无法覆盖的方面,以及规则。这些注解中,部分来自于多年技术工作者积累的文献中、高度内部优化的注解(注,这里指的,反复引用和论证的)。进一步,我们展示了这些模型(或论文)在以下场景的应用能力,即关于复合优化物中命中先导复合化合物,以及有偏的、从头学起的、机器学习输出的药物涉及结果。我们也展示了模型输出的评分函数,这些函数能够更准确地捕捉药物相似性,相较于另一种方式而言,即QED。我们进一步梳理了,模型输出的化学偏好,通过对大型复合型数据库进行分层分析。最后,为了促进再生产(论文的复制),以及促进其他相关主题研究,我们使用软件 MolSkill 进行对比,该软件包括准生产模型(production-ready model),匿名标记数据,我们通过许可的方式获得的数据。
lead optimization 先导化合物。药物化学术语。
wet-lab 湿法实验
play a central role 发挥核心作用
synthesize 化合物
ADMET 吸收(absorption)、分布(distribution)、代谢(metabolism)、排泄(excretion)和药代动力学(pharmacokinetics,PK)对功效和安全性都有显着影响,它们通常缩写为ADMET。来源:
知乎
compound prioritization 复合优先级
an in-house crowdsourced proxy for molecular complexity 分子复杂性的内部众包代理
porous organic cages 多空的有机物笼子。这里指的用于捕捉某种性质涉及的分子工具,ShoelessCai 认为。
cognitive bias 认知偏差
annotations 注解
silico chemoinformatics metrics 计算机化学信息度量
de novo 从头学起的
发现新药:“靶点”、“苗头化合物”、“先导化合物”
靶点 是一个宽泛的术语,它可以适用于一系列生物体,比如蛋白质、基因和RNA。一个好的靶点需要是有效的、安全的、临床可行且有商业价值的,最重要的是,要具有“可药性(druggable)”。
一旦选定了药物作用的靶点,药物化学家首先要找到对该靶点有作用的化合物。
苗头化合物 是指对特定靶标或作用环节具有初步活性的化合物。ShoelessCai 评注,类似于催化剂,药引之类。
一旦通过筛选或其他方式获得了大量的苗头化合物,药物发现团队的第一个任务就是确定哪些化合物是最好的研究对象。一般从多个苗头化合物中, 决策出活性最好的一个(或几个)作为
先导化合物 用于继续深入研究。
QED quantitative estimate of drug-likeness。是一种将药物相似性量化为介于0和1之间的数值的方法
03 结论
首先,我们关注一开始两个实验的评估结果,这些结果使得我们最终采取随后介绍的生产级别测试(production-level runs),在此之前,需要生产过程中预测表现模型输出一项定量评估结果。然后,我们开始探索几个领域,这些领域我们认为(我们开发的)评分函数是可行的。我们研究以下事物之间关系,包括模型输出的评分函数、其他常见的计算机化学度量方法,来评估这些方法能否区分不同性质的化学集合。进一步,我们考察模型能否输出更加精准的化学偏好,通过分层分析。最后,举例说明所提出的评分函数在有偏分子生成中的使用。
ShoelessCai评注,该文章对先导化合物阶段的化合物筛选,使用 Scoring 的方法助力识别候选及性质,并且与已有方法作比较。
chemical sets of different nature 不同性质的化学物集合
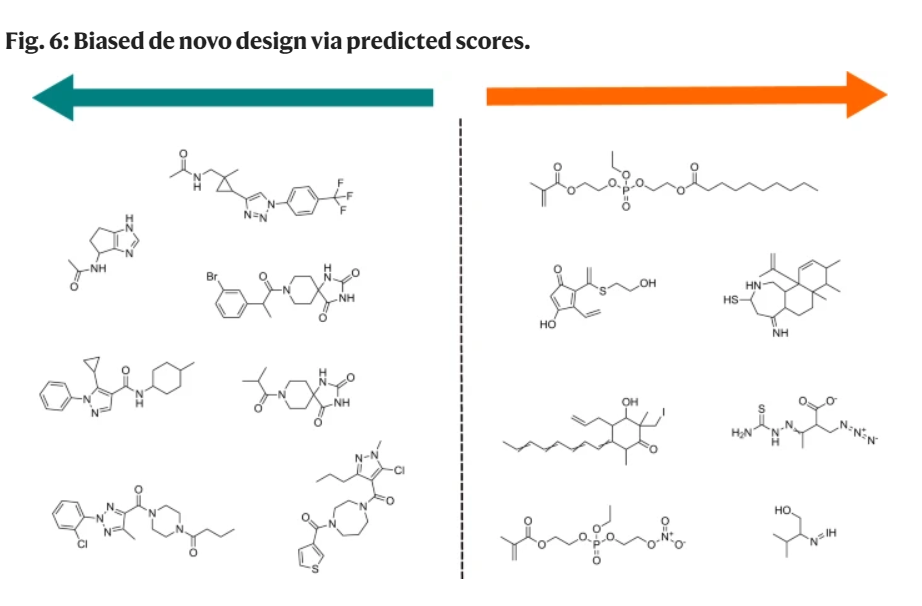
biased molecular generation 有偏分子生成
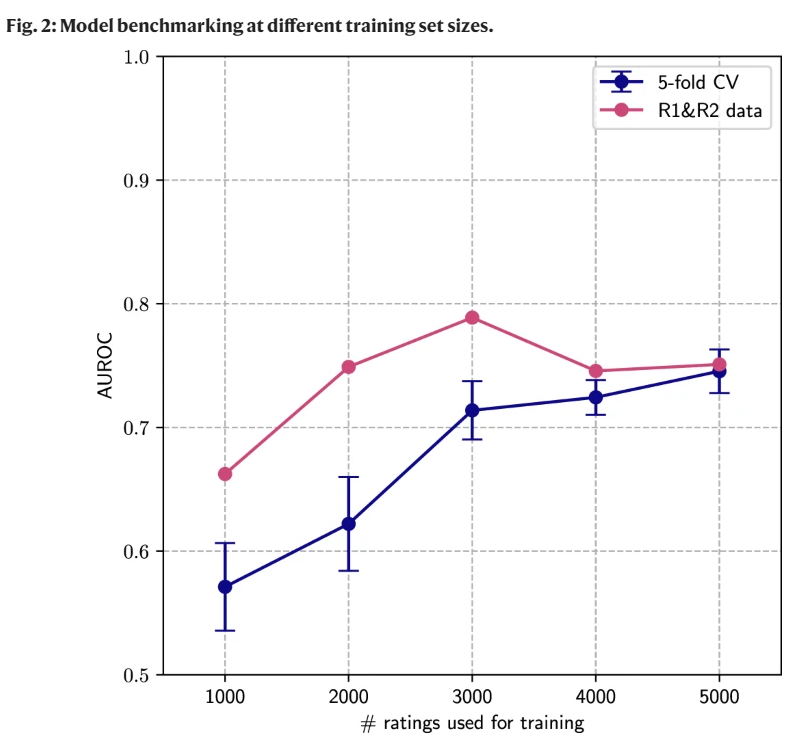
显然,用不同数据集效果明显好于 CV,但是样本量达到一定境界未必。
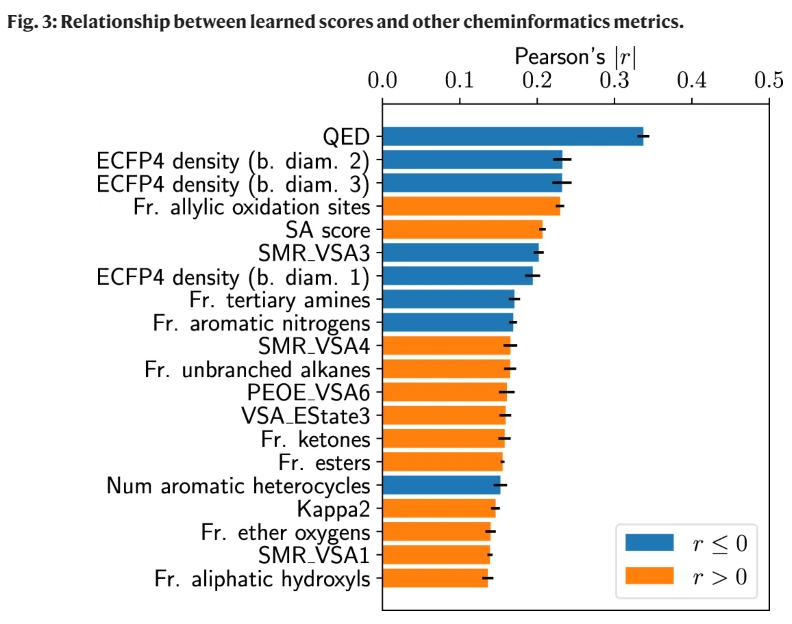
文章的神经网络方法,和QED 更具有线性相关性。
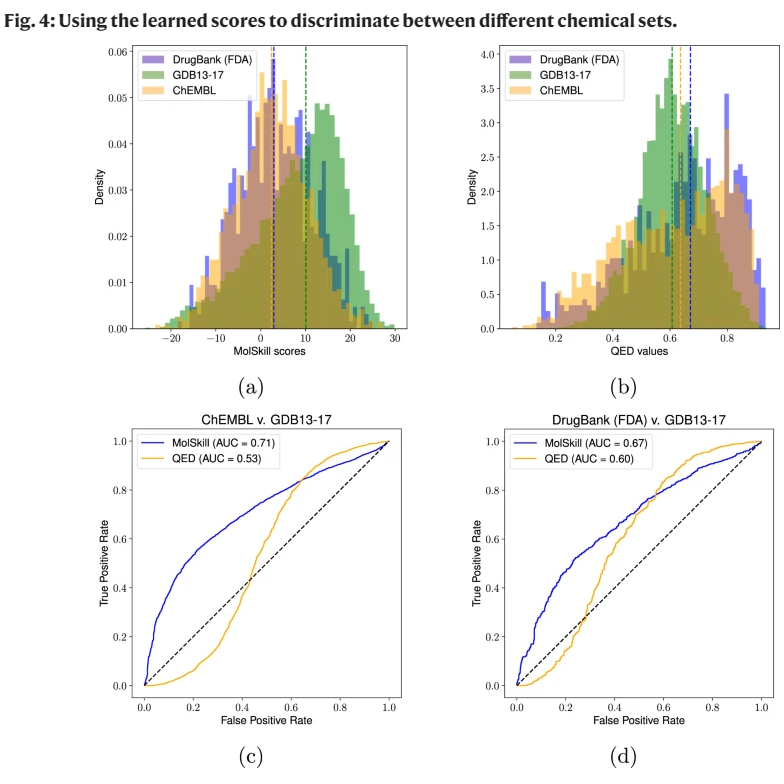
第一、二行两张图,主要考察 MolSkill 和 QED 差异,不难发现, MolSkill 有偏的,QED 是对称的
第三 ChEMBL v.s. GDB13-17 作为 0-1 建模之后的区分度;第四 DrugBank v.s. GDB13-17 区分。结果说明,某些方法是不是适应数据集,也决定了模型的区分程度。一般来说区分模型不太会呈现 S 型,QED 的图形,很可能是基于 QED 度量性质本身导致的。ShoelessCai 观点。
04 原文
Abstract
The lead optimization process in drug discovery campaigns is an arduous endeavour where the input of many medicinal chemists is weighed in order to reach a desired molecular property profile. Building the expertise to successfully drive such projects collaboratively is a very time-consuming process that typically spans many years within a chemist’s career. In this work we aim to replicate this process by applying artificial intelligence learning-to-rank techniques on feedback that was obtained from 35 chemists at Novartis over the course of several months. We exemplify the usefulness of the learned proxies in routine tasks such as compound prioritization, motif rationalization, and biased de novo drug design. Annotated response data is provided, and developed models and code made available through a permissive open-source license.
Introduction
Drug discovery is a complex, multi-step process that operates at the interface between many chemical and biological sub-disciplines. In many stages of the pipeline, and specifically during lead optimization, medicinal chemists—wet-lab or computational—play a central role, as they are routinely tasked with identifying which compounds to synthesize and evaluate over subsequent rounds of optimization1. In order to do this, medicinal chemists often review data that includes compound properties such as activity, ADMET, or target structural information, among many others. Therefore, for a campaign to be successful it needs not only rely on the quality of the generated experimental data, but ultimately also on the robustness and soundness of the decisions made by the medicinal chemistry team working on it.
During their professional careers, medicinal chemists build an expertise that enables them to make their decisions (e.g., compound prioritization) more efficiently4. That is, they develop an intuition on the factors relevant for a compound to be successful on following iterations of the early drug discovery process. While attempts have been previously made to formalize such knowledge with rule-based approaches (e.g., structural alerts), or simple cheminformatics desirability scores (e.g., drug-likeness), capturing the subtleties and intricacies involved in the ranking ability of chemists remains a fundamental challenge. With that motivation in mind, in this work we investigate whether part of this knowledge can be distilled into machine learning models. Such models can potentially then be deployed as an aid in during the decision-making process in lead optimization or other parts of the drug discovery pipeline, similar to other recommendation systems already reported in the industry.
Since medicinal chemistry is currently mostly a human endeavour, it is also inevitably prone to subjective biases8. Several studies have evaluated to what degree medicinal chemists tend to agree on their own and the decisions made by their colleagues. Most tasks explored in these works included presenting chemists with a list of compounds to filter over several rounds, in order to evaluate whether their choices overlapped with those of their peers, and if they were self-consistent with their own prior selections. These studies reported overall a weak agreement between and within each chemist—the disparity in these results being associated to several psychological factors, such as loss aversion11. Another study12, closer in nature to what we present in this work, evaluated whether a small group of chemists could rate compounds according to properties such as drug-likeness and synthetic accessibility via the use of a Likert-type scale13, to then train a classical machine learning model on the obtained responses. A more recent study by Merck14, used the same scaling strategy to model an in-house crowdsourced proxy for molecular complexity. While varying low to fair correlation degrees were found between the scores assigned by the chemists in the previous two studies, the reported study designs could have been prone to the anchoring psychological effect, in which decisions are affected by subject- and situation-specific reference values11. A recent work with a similar experimental setup was also described in the context of the design of porous organic cages.
In this study we set to overcome those limitations by adopting a strategy that is well-known in the context of multiplayer games. We cast the goal of ranking a set of molecules as a preference learning problem and show that individual preferences can be captured via pairwise comparisons with a simple neural network architecture. A basic schematic summarizing the idea behind the study is provided on Fig. 1. Proof-of-concept data collection rounds were carried out to evaluate whether the proposed study design and methodology successfully overcame cognitive bias limitations that were present in previous studies. 35 (wet-lab, computational, and analytical) chemists at Novartis participated in the study, with over 5000 annotations collected over several rounds driven by an active learning approach. We show that the learned implicit scoring functions capture aspects of chemistry currently not covered by other in silico chemoinformatics metrics and rule sets, some of them derived from highly optimised internal annotations over years of cumulative know-how. We furthermore exemplify their applicability in the context of hit-to-lead compound prioritization and biased de novo machine-learning drug design. We also show that the proposed learned scoring function can capture the concept of drug-likeness more accurately than another widely used metric (QED). We furthermore rationalize the learned chemical preferences by means of fragment analyses on a large public compound database. Finally, so as to facilitate reproducibility and foster additional research on this topic, a software package (MolSkill), containing production-ready models and anonymized response data, is made available through a permissive license in an accompanying code repository.
Result
We first focus on the evaluation of the results provided by two preliminary rounds for the study (see Methods), which ultimately led us to pursue the subsequent production-level runs. This is followed by a quantitative evaluation of predictive model performance over the production rounds. We then proceed to explore several areas where we believe the proposed scoring function can be practical. We study the relationship of the learned scoring function to other common in silico metrics in chemoinformatics and evaluate whether it can distinguish between chemical sets of different nature. We further investigate whether more precise learned chemical preferences can be rationalized via means of a fragment analysis and, finally, exemplify the usage of the proposed scoring function in biased molecular generation.